Kromozomlar gen formunda genetik bilgi içerir. Yumurta ve sperm dışında her insan hücresinin çekirdeği, 23 çift oluşturan 46 kromozom içerir. Her çiftteki bir kromozom anneden, diğeri ise babadan gelir. Her iki cinsiyette de 23 çift kromozomun 22'si aynıdır, yalnızca geri kalan cinsiyet kromozomu çifti farklıdır. Kadınlarda iki X kromozomu (XX), erkeklerde ise bir X ve bir Y kromozomu (XY) bulunur. Bu nedenle, bir erkek için normal kromozom seti (karyotip) 46, XY ve kadınlar için - 46, XX'dir.
Kromozom anormallikleri
Yumurta ve sperm üreten özel bir hücre bölünmesi sırasında bir hata meydana gelirse, anormal germ hücreleri ortaya çıkar ve bu da kromozomal anormalliğe sahip yavruların doğmasına yol açar. Kromozomal dengesizlik hem niceliksel hem de yapısal olabilir.
Çocuğun cinsiyet gelişimi
Normal şartlarda, Y kromozomunun varlığı, X kromozomu sayısına bakılmaksızın erkek fetüsün gelişmesine, Y kromozomunun yokluğu ise dişi fetüsün gelişmesine yol açar. Cinsiyet kromozomu anomalileri bireyin fiziksel özellikleri (fenotip) üzerinde otozomal kromozom anomalilerine göre daha az yıkıcı etkiye sahiptir. Y kromozomu az sayıda gen içerdiğinden ekstra kopyaların etkisi minimum düzeydedir. Hem erkekler hem de kadınlar yalnızca bir aktif X kromozomuna ihtiyaç duyar. Ekstra X kromozomları neredeyse her zaman tamamen aktif değildir. Bu mekanizma anormal X kromozomlarının etkisini en aza indirir, çünkü fazla ve yapısal olarak anormal kopyalar etkisiz hale getirilir ve yalnızca bir normal X kromozomu "çalışır" kalır. Ancak X kromozomunda inaktivasyonu önlemeyi başaran bazı genler vardır. Bu tür genlerin bir veya ikiden fazla kopyasının varlığının, cinsiyet kromozomu dengesizliğiyle ilişkili anormal fenotiplerden sorumlu olduğu düşünülmektedir. Laboratuvarda ışık mikroskobu altında 1000x büyütmede kromozom analizi yapılmaktadır. Kromozomlar ancak bir hücre genetik olarak aynı iki yavru hücreye bölündüğünde görünür hale gelir. Kromozom elde etmek için besin açısından zengin özel bir ortamda kültürlenen kan hücreleri kullanılır. Bölünmenin belirli bir aşamasında hücreler, kromozomların "çözülmesi" ve ayrılmasıyla birlikte şişmelerine neden olan bir çözelti ile muamele edilir. Hücreler daha sonra bir mikroskop lamı üzerine yerleştirilir. Kurudukça hücre zarı yırtılır ve kromozomlar dış ortama salınır. Kromozomlar, her birinin üzerinde sırası her çifte özel olan açık ve koyu diskler (şeritler) görünecek şekilde boyanır. Kromozomların şekli ve disklerin şekli, her bir kromozomu tanımlamak ve olası anormallikleri belirlemek için dikkatle incelenir. Kantitatif anormallikler, kromozom eksikliği veya fazlalığı olduğunda ortaya çıkar. Bu tür kusurlar sonucunda gelişen bazı sendromların belirgin belirtileri vardır; diğerleri neredeyse görünmezdir.
Her biri belirli bir sendromla ilişkili dört ana kantitatif kromozomal anormallik vardır: 45, X - Turner sendromu. 45,X veya ikinci cinsiyet kromozomunun yokluğu Turner sendromunda en sık görülen karyotiptir. Bu sendroma sahip bireyler kadındır; Hastalık genellikle ensedeki deri kıvrımları, el ve ayakların şişmesi ve düşük vücut ağırlığı gibi karakteristik belirtiler nedeniyle doğumda teşhis edilir. Diğer semptomlar arasında kısa boy, kanatlı kıvrımlı kısa boyun, geniş aralıklı meme uçları olan geniş bir göğüs, kalp kusurları ve önkolların anormal sapması yer alır. Turner sendromlu kadınların çoğu kısırdır, adet görmez ve ikincil cinsel özellikler, özellikle de meme bezleri gelişmez. Ancak hemen hemen tüm hastaların zihinsel gelişimi normal düzeydedir. Turner sendromunun görülme sıklığı kadınlarda 1:5000 ila 1:10.000 arasında değişmektedir.
■ 47, XXX - X kromozomunun trizomisi.
Yaklaşık 1000 kadından 1'inin karyotipi 47, XXX'tir. Bu sendroma sahip kadınlar genellikle uzun boylu ve zayıftır ve herhangi bir belirgin fiziksel anormallik yoktur. Ancak genellikle belirli öğrenme ve davranış sorunlarıyla birlikte IQ'larında düşüş yaşarlar. Trizomi X'li kadınların çoğu doğurgandır ve normal sayıda kromozoma sahip çocuk sahibi olabilirler. Fenotipik belirtilerin hafif şiddeti nedeniyle sendrom nadiren tespit edilir.
■ 47, XXY - Klinefelter sendromu. Yaklaşık 1000 erkekten 1'inde Klinefelter sendromu var. 47,XXY karyotipine sahip erkekler, küçük öğrenme ve davranış sorunları dışında doğumda ve erken çocukluk döneminde normal görünürler. Karakteristik belirtiler ergenlik döneminde fark edilir hale gelir ve bunlar arasında uzun boy, küçük testisler, sperm eksikliği ve bazen genişlemiş meme bezleri ile birlikte ikincil cinsel özelliklerin zayıf gelişimi yer alır.
■ 47, XYY - XYY sendromu. Yaklaşık 1000 erkekten 1'inde fazladan bir Y kromozomu bulunur. XYY sendromlu erkeklerin çoğu görünüşte normal görünür ancak çok uzundurlar ve zeka düzeyi düşüktür. Kromozomlar belli belirsiz X harfine benzer şekildedir ve iki kısa ve iki uzun kolu vardır. Aşağıdaki anomaliler Turner sendromu için tipiktir: uzun kol boyunca izokromozom. Yumurta veya sperm oluşumu sırasında kromozomlar ayrılır ve ayrışma bozulursa, iki uzun kollu ve kısa kolların tamamen bulunmadığı bir kromozom ortaya çıkabilir; halka kromozomu. X kromozomunun kısa ve uzun kollarının uçlarının kaybedilmesi ve kalan bölümlerin bir halka halinde birleşmesi sonucu oluşur; X kromozomlarından birinin kısa kolunun bir kısmının silinmesi (kaybı). X kromozomunun uzun kolundaki anormallikler genellikle erken menopoz gibi üreme sistemi işlev bozukluklarına neden olur.
Cinsiyet kromozomlarının patolojileri, sayılarının ihlali (anöploidi) veya yapısal kusurlardan kaynaklanabilir.
En yaygın cinsiyet kromozomu anöploidileri şunlardır: 45.X (Turner sendromu); 47,XXY (Klinefelter sendromu); 47,XYY; ve 47,XXX. Normal genotipli hücrelerin vücudunda bulunmasıyla birlikte cinsiyet kromozomlarının mozaiği nadir değildir. Cinsiyet kromozomu mozaikçiliğinin en yaygın iki türü 45,X/46,XX ve 45,X/46,XY'dir. Mozaikizmli hastalarda fenotipik belirtilerin ciddiyeti, anormal hücrelerin oranına karşılık gelir.
X ve Y kromozomlarının yapısal patolojileri öncelikle izokromozomları, delesyonları, duplikasyonları, halka kromozomlarını ve translokasyonları içerir.
Genomik bozukluğun bir örneği gen kopyalanmasıdır MECP2 erkeklerde kas hipotonisi, şiddetli zihinsel gerilik, gecikmiş konuşma gelişimi, yutma bozuklukları, sık görülen solunum yolu enfeksiyonları ve ayrıca konvülsif nöbetler (tedavi edilemeyen tonik-klonik nöbetler) varlığında ifade edilir.
Kromozom sayısındaki anormallikler (anöploidi)
En yaygın cinsiyet kromozomu anöploidileri 45.X'tir (Shereshevsky-Turner sendromu); 47,XXY (Klinefelter sendromu); 47,XYY ve 47,XXX görülme sıklığı sırasıyla yaklaşık 1/2500, 1/500 ila 1/1000, 1/900 ila 1500 ve 1/1000'dir. Normal genotipli hücrelerin vücudunda bulunmasıyla birlikte cinsiyet kromozomlarının mozaiği nadir değildir. Cinsiyet kromozomu mozaikçiliğinin en yaygın iki türü 45,X/46,XX ve 45,X/46,XY'dir. Mozaikizmli hastalarda fenotipik belirtilerin ciddiyeti, anormal hücrelerin yüzdesine karşılık gelir.
X kromozomunda monozomi (45.X veya Shereshevsky-Turner sendromu)
Shereshevsky-Turner sendromlu hastaların çoğunda X kromozomu karyotip 45.X'te monozomi vardır. Sendromun diğer formları, Y kromozomunun kısmen silinmesiyle birlikte 45,X/46,XX veya 45,X/46,XY gibi X kromozomunda mozaikçiliği içerir. Bazı hastalarda ikinci X kromozomunda yapısal bir anormallik vardır (örneğin, X kromozomunun uzun kol izokromozomisi veya kısa kolun silinmesi). Y kromozomunun kısa kolunun distal kısmını içeren delesyonlar da Turner sendromu fenotipiyle ilişkilidir, çünkü bu durumda hastalarda anti-Turner genleri (SHOX, RPSY4 ve ZFY) yoktur. X kromozomunun kısa kolunun silinmesi de Turner sendromu fenotipiyle ilişkilendirilmiştir. Çoğu izole vakalardır.
Shereshevsky-Turner sendromu boy kısalığı ve aşağıdakilerden bazılarıyla karakterizedir: düşük kulaklar dahil yüz dismorfisi, boyunda deri kıvrımları, kalkan şeklinde göğüs (geniş, meme uçları arasında geniş mesafe), lenfödem, valgus deformitesi dirsek eklemi, kısa dördüncü metakarpal kemik, tırnak plakalarının hipoplazisi, yaşlılık lekeleri ve konjenital kalp defektleri. Kalp kusurları arasında tipik ve en yaygın olanı damar defektleri ve aort koarktasyonudur. Ayrıca Turner sendromundan muzdarip hastalarda çizgi benzeri gonadlar gelişir, yumurtlama bozukluğu olur ve cinsel gelişimde gecikme olur. Böbrek gelişimindeki bozukluklar (at nalı böbrek) de ortaya çıkar. Alt ekstremite lenfödemi yenidoğanlarda görülen tek klinik belirti olabilir. Y kromozomundan genetik materyal taşıyan Turner sendromlu bireylerde gonadoblastoma gelişme riski yüksektir.
47.XXY Klinefelter sendromu
Klinefelter sendromu, primer hipogonadizme neden olan cinsiyet kromozomlarının sayısının en sık görülen patolojisidir. 47,XXY karyotipi cinsiyet kromozomunun ayrılmamasının bir sonucudur ve anne ya da baba kökenli olabilir. Hastalık vakalarının çoğu doğumdan sonra tespit edilir ve kısırlığın nedenleri belirlenirken, jinekomasti, kriptorşidizm veya nörolojik bozuklukların tanımlanması sırasında teşhis edilir.
Pirinç. Cinsiyet kromozomlarının ayrılmaması
47,XXY karyotipli yeni doğan erkek çocuklar fenotipik olarak normaldir, fizyolojik olarak normal erkek dış cinsel organlarına sahiptir ve herhangi bir görünür dismorfi yoktur. Klinefelter sendromunun uzun boy, küçük testisler ve kısırlık (azospermi) dahil olmak üzere ana klinik belirtileri ergenlik sonrası dönemde belirgin hale gelir. Klinefelter sendromlu hastalarda zihinsel bozukluklar, otizm spektrum bozuklukları ve sosyal sorunlar açısından artmış risk vardır. Klinefelter sendromu tanısı alan hastaların nörolojik durumlarının değerlendirilmesi ve bir endokrinoloğa yönlendirilmesi gerekir.
47.XYY
47,XYY karyotipine sahip bireyler uzun boyludur ve motor ve konuşma gelişiminde orta derecede gecikmeler yaşayabilirler. Birçoğu öğrenmeye daha fazla dikkat gerektiriyor, ancak kural olarak hepsi temel kapsamlı okullarda okuyor. Ergenlik gelişimi normaldir ve çoğu erkek çocuk doğurgandır. Fenotipin hafif olması ve buna bağlı herhangi bir sağlık sorunu olmaması nedeniyle, 47,XYY karyotipine sahip birçok kişiye yaşamları boyunca teşhis konulmadan kalır.
Daha önce 47.XYY'ye sahip erkeklerin saldırganlıklarının arttığı ve bunun da saldırgan davranışlarına yansıdığı bildirilmişti. Bununla birlikte, Avrupalı ve Amerikalı genetikçilerin daha sonraki büyük ölçekli ortak çalışmaları, XYY'li erkeklerin artan suç faaliyetlerine ilişkin istatistiklerin, düşük IQ'ya (yaklaşık 10 puan) bağlı olarak düşük sosyo-ekonomik statüleri ile ilişkili olduğunu gösterdi; bu da, öğrenmede bazı zorluklara yol açtı. kanun ve daha sıklıkla küçük suçlar. 47,XYY'li bireylerde dikkat eksikliği hiperaktivite bozukluğu ve otizm spektrum bozukluğu oranları daha yüksektir. Öğrenme güçlüğü ve davranış sorunlarının yaygınlığı göz önüne alındığında, bu hastalar için nörogelişimsel değerlendirme önerilmektedir.
47.XXX
47,XXX (trizomi X olarak da bilinir), kadınlarda cinsiyet kromozomlarının en sık görülen patolojisidir. Trizomi X tanısı anne karnında genetik tarama sırasında konur. 47.XXX karyotipi olan kadınların kromozomal anormallikleri olan bir fetüs geliştirme riski artmamaktadır.
Karyotipi 47.XXX olan 155 kadın üzerinde yapılan bir araştırma, bunların yüzde 62'sinin fiziksel olarak normal olduğunu gösterdi. Bu nedenle 47.XXX karyotipi olan çoğu kişiye hiçbir zaman teşhis konulamaz. 47.XXX'li kadınların büyümesi yüksek; (ortalama baş çevresi 25. ve 35. yüzdelik dilim arasında değişir, ancak çoğu kişide ergenlik döneminde 80. yüzdelik dilime ulaşabilir). Cinsel olgunluk ve doğurganlık çoğunlukla normaldir ancak erken yumurtalık yetmezliği meydana gelebilir.
Karyotipi 47.XXX olan on bir bebek üzerinde yapılan daha sonraki bir çalışma, kızların doğumdan itibaren IQ'larının erkek kardeşlerininkinden 15-20 puan daha düşük olduğunu gösterdi. Bu nedenle gelişimsel gecikmelerin takip edilmesi ve ileride psikolojik sorunların varlığının tespit edilmesi önerilmektedir.
Diğer hastalıklar
Yüzden fazla 49,XXXXY karyotipi vakası rapor edilmiştir; en az yirmi vaka 49,XXXXX ve birkaç vaka 49,XYYYY'dir. Ek cinsiyet kromozomlarının sayısı ile hastalarda fenotipik belirtilerin şiddeti arasında doğrudan bir ilişki vardır. Cinsiyet kromozomlarının tetra ve pentazomisi üzerine yapılan bir çalışma, X kromozomu üzerindeki polisominin, Y kromozomu üzerindeki polisomiden daha ciddi sonuçlarla ilişkili olduğu sonucuna varmıştır. IQ seviyelerinin her ekstra X kromozomu için normal sayıdan 10 puan azaldığı gösterilmiştir.
49.XXXXY XXXXY karyotipinin karakteristik klinik özellikleri geniş veya kalkık burun ucuyla birlikte çökmüş burun köprüsü, geniş aralıklı gözler, göz kapağı-burun kıvrımları, iskelet patolojileri (özellikle radyoulnar sinostoz), konjenital kalp hastalıkları, endokrin bozuklukları ve yüksek derecede Hipogonadizm ve hipogenitalizm. Şiddetli zeka geriliği ve orta derecede boy kısalığı da yaygındır. Bu karyotipe sahip bireyler sıklıkla Klinefelter sendromu vakaları olarak sınıflandırılsa da, XXXXY'nin tüm karakteristik özellikleri oldukça açık bir şekilde bu fenotipe işaret etmektedir.
49,XXXXX Karyotipi 49,XXXXXX (X kromozomu pentazomisi) olan kadınlarda her zaman zeka geriliği vardır. Kraniofasiyal, kardiyovasküler ve iskelet patolojileri gibi diğer bulgular oldukça değişkendir. Pentazomi X'li hastalar Down sendromunda görülenlere benzer özellikler sergileyebilir. Radioulnar sinostoz, çok sayıda X kromozomu olan hastalarda da yaygındır. Bazı hastalarda 48,XXXX ve 49,XXXXX mozaikçiliği vardır.
Mozaiklik 45,X/46,XX
Bu en yaygın cinsiyet kromozomu mozaikçiliğidir ve amniyosentez ve doğum öncesi karyotipleme ile teşhis edilir. Bu tür mozaikçiliğe sahip bireyler Turner sendromunun daha hafif klinik özelliklerine sahiptir. Pek çok kadın ergenlik dönemini geçirdi ve üremeyi başardı.
Doğum öncesi teşhis edilen 156 45.X/46.XX mozaikçilik vakasının %14'ünde anormal sonuç elde edildi. İki ölü doğum ve 20 anormal fenotip vakası vardı (12'sinde Turner sendromunun bazı özellikleri vardı ve geri kalan 8'i anormaldi, muhtemelen ilgisizdi). Kızların yüzde 85'inden fazlasının doğumda normal bir fenotipi vardı ya da bu durum hamileliğin tıbbi olarak sonlandırılması sonucu oluşmuştu. Ancak Turner sendromunun temel özellikleri (boy kısalığı ve ikincil cinsel özelliklerin yokluğu gibi) yalnızca çocukluk veya ergenlik döneminde ortaya çıkar ve bebeklik döneminde fark edilmez. Normal fenotipe sahip, yumurtalık fonksiyonu bozulmuş bazı kadınlarda 45,X/46,XX mozaikliği tespit edilir.
Mozaiklik 45,X/46,XY
45,X/46,XY'nin varlığıyla oluşan mozaiklik geniş bir fenotipik spektruma sahiptir. Örneğin, doğum sonrası teşhis edilen 151 45,X/46,XY mozaikçiliği vakasından oluşan retrospektif bir seride, hastaların %42'si fenotip olarak kadındı ve tipik veya atipik Turner sendromu mevcuttu. Diğer bir %42'nin ise belirsiz dış cinsel organları ve asimetrik gonadları (karışık gonadal disgenezi) vardı ve son olarak %15'inde tamamlanmamış maskülinizasyona sahip bir erkek fenotipi vardı. Dolayısıyla doğum sonrası teşhis edilen tüm vakalar fenotipik olarak anormaldi. Buna karşılık, doğum öncesi teşhis edilen 80 45,X/46,XY 74 mozaikçilik vakasının %92,6'sı fenotipik olarak normal erkek çocuklardı. Bu, mozaikçiliğe sahip ancak normal fenotipe sahip çocukların veya yetişkinlerin tıbbi yardım arama ihtimalinin düşük olduğu (sevk yanlılığı) gerçeğini açıklayabilir.
Kromozomların yapısal anormallikleri
Yapısal patolojiler öncelikle izokromozomları, delesyonları, duplikasyonları, halka kromozomlarını ve translokasyonları içerir.
İzokromozom Xq
Kısa kolun (p) elimine edildiği (yok/azaltılmış) ve uzun kolun (q) tam bir kopyasıyla değiştirildiği X kromozomunun uzun kolunun izokromozomu, isoXq veya i(Xq), en yaygın olanıdır cinsiyet kromozomlarının anormalliği.
Yapısal patolojinin varlığı ebeveynlerde yaşa bağlı risk artışıyla ilişkili değildir. İzokromozomi 46,X,i(Xq), vücutta genetik olarak farklı iki hücre popülasyonu mevcut olduğunda mozaikçiliğin bir ifadesi olabilir: normal - 46,XX ve 45,X.
Xq ve Xy izokromozomları Turner sendromuyla ilişkilidir, bunun nedeni muhtemelen ana anti-Turner geni SHOX'un X ve Y kromozomlarının kısa kollarının distal kısmında (psödootozomal bölgelerde) bulunmasıdır. İzokromozom Xq, Klinefelter sendromunun varyasyonlarından biri olan 47,X,i(Xq),Y'ye sahip hastalarda da tespit edilir.
Xp22.11 silme
Xp22.11 silinmesi geni içerir PTCHD1. Otistik bozukluğu olan birkaç ailede ve zihinsel engelli üç ailede kimliklendirme bildirildi. Gen PTCHD1 otizmli veya otizmsiz X'e bağlı zeka geriliği için aday bir gendir. Bu genin işlevi ve rolü bilinmemektedir.
Xp22.3 silme
Bu bölgenin silinmesi sıklıkla mikroftalmi ve lineer cilt kusurları sendromu (MLS) ile ilişkilidir ve X'e bağlı dominant bir hastalıktır, yani erkeklerde öldürücüdür ve bu nedenle sadece kadınlarda görülür. Bu bölgedeki gen mitokondriyal sitokrom c sentazını kodlar ( HCCS). MLS'nin klinik belirtisi, mikroftalmi ve anoftalmi (tek taraflı veya iki taraflı) ve çoğunlukla yüz ve boyunda zamanla düzelen doğrusal cilt defektlerinin varlığı ile ifade edilir. Beynin yapısal patolojileri, gelişimsel gecikmeler ve nöbetler (nöbetler) de klinik tablonun bir parçasıdır. Kardiyak anormallikler (hipertansif kardiyomiyopati ve aritmi gibi), kısa boy, hiatal herni, tırnak distrofisi, preauriküler fistül, işitme kaybı, genitoüriner malformasyonlar (malformasyonlar, malformasyon) da yaygın klinik fenomenlerdir.
Tarama değerlendirmesi oftalmolojik ve dermatolojik muayeneyi, genel gelişimin değerlendirilmesini, ekokardiyogramı, beyin manyetik rezonans görüntülemesini (MRI) ve elektroensefalogramı (EEG) içerir.
Xp22 SHOX silme işlemleri
Xp22 delesyonu, idiyopatik boy kısalığına neden olan bir mutasyon olan SHOX genini içerir. SHOX geni X ve Y kromozomlarının psödootozomal 1. bölgesinde bulunur. Bu genin Turner sendromunda boy kısalığından sorumlu olduğuna inanılmaktadır ve bu genin haploins yetmezliği Lery-Weillian diskondrosteozuna neden olmaktadır. Lery-Weill'in diskondrosteozu, en çok kadınlarda belirgin olan kısa boy ve elin kronik subluksasyonu (bilek kemiklerinin deformasyonu, Madelung deformitesi) ile karakterizedir. SHOX geninin homozigot silinmesi, metafiz displazisinin daha şiddetli bir formu olan Langer displazisine neden olur. İskelet yapısında başka bir spesifik özellik bulunmayan, boy kısalığı olan hastalarda SHOX gen delesyonları kolaylıkla tespit edilir. SHOX yeniden düzenlemelerinin %60'ından fazlası gen silmeleridir; delesyonların yokluğunda, karşılaştırmalı genomik hibridizasyon ve ardından nokta mutasyonlarını tanımlamak ve oluşturmak için sıralama, idiyopatik kısa boy için klinik bir araştırmadır.
Xp11.22 silme işlemleri
Xp11.22 bölgesinin silinmesi, mutasyonları zihinsel gerilik, yarık dudak/damak ve otistik bozukluklarla ilişkilendirilen PHF8 genini (PHD8 parmak proteinini kodlayan) içerir.
PHF8 geninin silinmesine neden olan mutasyonlar, X'e bağlı zeka geriliği sendromu, Siderius-Hamel sendromu ile ilişkilidir.
Xp.22.31 kopyaları
Xp.22.31 lokusundaki kopyalar literatürde sıklıkla tanımlanmaktadır. Gen kopya sayısı değişiminin sonuçlarını belirlemenin zorluğu göz önüne alındığında, bu kopyalamanın patojenik mi yoksa iyi huylu mu olduğu konusunda pek çok tartışma olmuştur. Bu kopyalama steroid sülfataz genini etkiler. Sonuç, steroid sülfataz geninde, aktivitesinde bir azalma veya sentezinin yokluğu ile ifade edilen bir mutasyon olan genetik bir kusurdur. Bu genin silinmesi erkeklerde X'e bağlı iktiyoz ile ilişkilidir. Bu kopyalanma zeka geriliği olan hastalarda görülür. Ancak bu hastaların hem sağlıklı akrabalarında hem de genel popülasyonda tespit edilmektedir. Bu genin kopyaları fenotipik belirtilere sahip olmasa da, üçlü kopyalar sürekli olarak zihinsel bozukluklarla ilişkilendirilmiştir. FISH teşhisi sonuçta kopyaları üçlemelerden ayırt etmeyi (bir genin kopya sayısındaki artışı tanımak için) mümkün kılar.
ME2CP çoğaltma sendromu
Metil bağlayıcı CpG terminal proteini 2'yi kodlayan gendeki mutasyonlar ( ME2CP), Xq28'de bulunur ve Rett sendromundan sorumludur. Bu bölgenin çoğalmasının kadınlarda fenotipik önemi çok azdır veya hiç yoktur, bunun nedeni muhtemelen patolojik X kromozomunun inaktivasyonudur. Bu mutasyona sahip erkekler büyük ölçüde zayıflar. Çoğalmanın varlığı klinik olarak şiddetli kas hipotonisi, şiddetli zihinsel gerilik, gecikmiş konuşma gelişimi, yutma bozuklukları (yeme zorluğu), sık görülen solunum yolu enfeksiyonları ve bazen tedavi edilemeyen tonik-klonik olanlar da dahil olmak üzere konvülsif nöbetlerin varlığında ifade edilir. Bu kopyanın olduğu birçok hastaya otizm veya benzer tipte bir bozukluk tanısı konuldu. Rett sendromunda görülene benzer şekilde, duplikasyonu olan hastalarda ME2CP gelişimsel gerileme yaşıyor. Ayrıca ataksi gelişir ve vücudun alt kısmındaki ilerleyici kas spastisitesi sıklıkla yürüme kaybına yol açar. Gastrointestinal problemler ve şiddetli kabızlık kaydedildi. Çoğaltma sıklıkla interlökin 1 reseptör antagonist genini etkiler ( IRAK1), bu grup hastalarda gözlenen immün patolojilerin ortaya çıkmasında rol oynayabilir. Prognoz kötüdür ve bu kopyaya sahip erkeklerin çoğu, ikincil solunum yolu enfeksiyonları nedeniyle 30 yaşından önce ölmektedir. Bu bölgenin üçlenmesi erkeklerde daha da şiddetli bir fenotiple sonuçlanır.
Bu hastaların tarama muayeneleri arasında EEG, yutma fonksiyonunun değerlendirilmesi, humoral ve hücresel bağışıklığın değerlendirilmesi yer alır. Tedavi, kas hipotonisi ve spastisite tedavisini, konuşma terapisini (konuşma terapisi), beslenme sorunları için gastrostomi tüpünün kullanımını ve solunum yolu enfeksiyonlarının tedavisini içerebilir.
UpTodate web sitesindeki materyallerin çevirisi İmmünoloji ve Üreme Merkezi uzmanları tarafından hazırlandı.
Kromozomlar boydan göz rengine kadar her şeyle ilgili kalıtsal bilgileri taşıyan iplik benzeri moleküllerdir. Bunlar proteinden ve organizmanın ebeveynlerinden aktarılan genetik talimatlarını içeren tek bir DNA molekülünden oluşur. İnsanlarda, hayvanlarda ve bitkilerde kromozomların çoğu hücre çekirdeğinde çiftler halinde düzenlenmiştir. İnsanlarda otozom adı verilen bu kromozom çiftlerinden 22 tanesi bulunur.
İnsanlarda 22 çift kromozom ve iki cinsiyet kromozomu vardır. Kadınların iki X kromozomu vardır; erkeklerde bir X kromozomu ve bir Y kromozomu bulunur.
Cinsiyet nasıl belirlenir?
İnsanlarda toplam 46 kromozom olmak üzere fazladan bir çift cinsiyet kromozomu vardır. Cinsiyet kromozomlarına X ve Y adı verilir ve bunların kombinasyonu kişinin cinsiyetini belirler. Tipik olarak kadınlarda iki X kromozomu, erkeklerde ise XY kromozomu bulunur. Bu XY cinsiyet belirleme sistemi çoğu memelinin yanı sıra bazı sürüngenler ve bitkilerde de bulunur.
XX veya XY kromozomlarının varlığı, spermin bir yumurtayı döllemesiyle belirlenir. Vücuttaki diğer hücrelerin aksine, yumurta ve spermdeki gamet veya cinsiyet hücreleri adı verilen hücrelerde yalnızca bir kromozom bulunur. Gametler mayotik hücre bölünmesiyle üretilir; bu, bölünmüş hücrelerin ebeveynler veya atalar olarak kromozom sayısının yarısına sahip olmasıyla sonuçlanır. İnsanlarda bu, ana hücrelerin iki kromozoma ve bir gamete sahip olduğu anlamına gelir.
Annenin yumurtalarındaki tüm gametler X kromozomuna sahiptir. Babanın sperminde yaklaşık yarım X ve yarım Y kromozomu bulunur. Sperm çocuğun cinsiyetini belirleyen değişken bir faktördür. Sperm X kromozomu taşıyorsa yumurtanın X kromozomu ile birleşerek dişi zigot oluşturur. Sperm Y kromozomunu taşıyorsa erkek çocuk doğar.
Döllenme sırasında spermden gelen gametler yumurtadan gelen gametlerle birleşerek bir zigot oluşturur. Zigot, gerekli 46 kromozom için iki set 23 kromozom içerir. Dünya Sağlık Örgütü'ne göre çoğu kadın 46XX ve çoğu erkek 46XY'dir.
Ancak bazı seçenekler var. Son araştırmalar, bir kişinin, özellikle de kendisini LGBT olarak tanımlayanların birçok farklı cinsiyet kromozomu ve gen kombinasyonuna sahip olabileceğini gösterdi. Örneğin, Psikolojik Tıp dergisinde 2014 yılında yapılan bir araştırmaya göre, Xq28 adı verilen belirli bir X kromozomu ve kromozom 8 üzerindeki bir gen, eşcinsel erkeklerde daha yüksek oranda bulunuyor gibi görünüyor.
Bin bebekten birkaçı bir cinsiyet kromozomuyla (45X veya 45Y) doğar, buna monozomi denir. Diğerleri üç veya daha fazla cinsiyet kromozomuyla (47XXX, 47XYY veya 47XXY, vb.) doğarlar, buna polisomi denir. WHO, "Ayrıca bazı erkekler, Y kromozomunun cinsiyeti belirleyen bölgesinin küçük bir kısmının yer değiştirmesi nedeniyle 46XX ile doğuyor" diyor. “Benzer şekilde bazı kadınlar da Y kromozomundaki mutasyonlar nedeniyle 46XY olarak doğuyorlar. XX olanların sadece kadınlar ve XY olan erkeklerin olmadığı, bunun yerine bir dizi kromozom ilavesinin, hormonal dengelerin ve fenotipik varyasyonların olduğu açıktır."
X ve Y kromozomlarının yapısı
Vücudun diğer bölgelerine ait kromozomlar aynı boyut ve şekle sahip olup aynı eşleşmeyi oluştururken, X ve Y kromozomları farklı yapılara sahiptir.
X kromozomu, Y kromozomundan önemli ölçüde daha uzundur ve yüzlerce gen daha içerir. X kromozomundaki ek genlerin Y kromozomunda karşılığı olmadığından X genleri baskındır. Bu, X üzerindeki hemen hemen her genin, dişide resesif olsa bile, erkeklerde ifade edileceği anlamına gelir. Bunlara X'e bağlı genler denir. Yalnızca Y kromozomunda bulunan genlere Y bağlantılı genler adı verilir ve yalnızca erkeklerde ifade edilir. Herhangi bir cinsiyet kromozomundaki genlere cinsiyet genleri denilebilir.
Yaklaşık 1.098 X'e bağlı gen vardır, ancak bunların çoğu dişi anatomik özelliklerine yönelik değildir. Aslında bunların çoğu hemofili, Duchenne kas distrofisi ve diğer bazı bozukluklarla ilişkilidir. En sık erkeklerde bulunurlar. X'e bağlı genlerin cinsiyet dışı özellikleri de erkek tipi kellikten sorumludur.
Büyük X kromozomunun aksine Y kromozomu yalnızca 26 gen içerir. Bu genlerin on altısı hücre bakımından sorumludur. Dokuz tanesi sperm üretiminde rol oynar ve bazıları eksik veya kusurluysa, düşük sperm sayısı veya kısırlık meydana gelebilir. SRY geni adı verilen bir gen, erkeğin cinsiyet özelliklerinden sorumludur. SRY geni, Sox9 adı verilen cinsiyet dışı bir kromozomda bulunan başka bir genin aktivasyonunu ve düzenlenmesini tetikler. Sox9, cinsel olmayan gonadların yumurtalıklar yerine testislere doğru gelişmesini tetikler.
Cinsiyet kromozomu bozuklukları
Cinsiyet kromozomlarının kombinasyonundaki anormallikler, nadiren ölümcül olan çeşitli cinsiyete özgü koşullara yol açabilir.
Kadın anormallikleri Turner sendromuna veya Trizomi X'e yol açar. Turner sendromu, kadınlarda iki yerine yalnızca bir X kromozomuna sahip olduğunda ortaya çıkar. Semptomlar arasında cinsel organların normal şekilde olgunlaşmaması, kısırlığa, küçük göğüslere ve adet görmeme gibi durumlara yol açabileceği; kısa boy; geniş, tiroid göğsü; ve geniş bir boyun.
Trizomi X sendromuna iki yerine üç X kromozomu neden olur. Semptomlar arasında uzun boy, konuşma gecikmeleri, erken yumurtalık yetmezliği veya yumurtalık anormallikleri ve zayıf kas tonusu yer alır; ancak birçok kız ve kadın hiçbir belirti göstermez.
Klinefelter sendromu erkekleri etkileyebilir. Semptomlar arasında meme gelişimi, büyük kalçalar gibi anormal oranlar, uzun boy, kısırlık ve küçük testisler yer alır.
Erkek Y kromozomu
Kısa bilgi (video, İngilizce): ,
Kadın ve erkeklerin her birinde 23 çift kromozom bulunur. Her çiftten biri babadan, biri anneden alındı. “1”den “22”ye kadar sırayla isimlendirilen otozomal kromozomların aksine, iki “cinsiyet” kromozomunun harf isimleri vardır. Kadınlar için XX, erkekler için XY. Anneden - her zaman X kromozomu. Babadan çocuğa ya X kromozomu (kız) ya da Y kromozomu (erkek) miras kalacaktır. Babadan gelen X kromozomu XX kombinasyonuna dönüşür ve bu kadın cinsiyetidir. Babadan gelen Y kromozomu XY kombinasyonuna dönüşerek erkek cinsiyetini belirler. Hemen hemen tüm kromozomlar, her bir kromozom çiftinin birbiriyle farklı parçalar değiştirdiği bir süreç olan karıştırmaya (rekombinasyon) tabi tutulur. Her insanda yalnızca bir Y kromozomu bulunduğundan, X kromozomlarından farklı olarak yeniden birleşmez. Bu nedenlerden dolayı X kromozomları üzerinde yapılan soy analizi çok daha karmaşık hale gelmektedir. Ayrıca mitokondriyal DNA'yı (mtDNA) annemizden alırız, ancak babamızdan almazız.
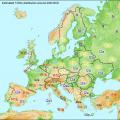
DNA şeceresinin ana araçları mutasyonların, bunların sayısının ve mtDNA ve Y kromozomlarındaki konumlarının analizidir. Y kromozomu, mitokondriyal DNA'nın aksine, çok düşük mutasyon sıklığı ve karışım (rekombinasyon) olmaması nedeniyle nesilden nesile neredeyse hiç değişmeden aktarılır. Mutasyon varyasyonlarına bağlı olarak kromozomlar haplotiplere bölünür ve bunlar haplogruplar ve alt gruplar (alt gruplar) halinde birleştirilir. Haplogrupların harf tanımları alfabetiktir ve bir sonraki mutasyonun ortaya çıkma zamanını gösterir. Yani, haplogrup A (yaklaşık 75.000 yıl önce ortaya çıkan, bugün esas olarak Güney Afrika'da lokalize olan sözde Adem'in Y kromozomu) yaş olarak daha yaşlıdır (yaklaşık 30.000 yıl önce), vb. alfabetik.
Y-DNA haplogruplarının tahmini dağılımı MÖ 2000. e.

Y-DNA haplogruplarının dağılımı

Y-DNA haplogruplarının Avrupa'daki dağılımı
Bildiğiniz gibi erkek hücrelerinde bir çift cinsiyet kromozomu bulunur: dişi X kromozomu ve erkek Y kromozomu. Bununla birlikte, eğer Y kromozomu gerçekten erkekse, o zaman X kromozomu oldukça yaygındır: sonuçta her iki cinsiyetin temsilcileri de buna sahiptir, ancak bunu annelerinden alan erkeklerde yalnızca bir kopyada bulunur. X kromozomu, cinsiyete bakılmaksızın herhangi bir organizmanın yaşamı için gerekli olan birçok geni içerir. Aynı zamanda hemofili, Duchenne kas distrofisi ve renk körlüğü gibi ciddi hastalıklara yol açan genleri, mutasyonları da içerir. X'e bağlı bu hastalıklar çoğunlukla erkekleri etkiler çünkü onlarda yalnızca bir X kromozomu vardır.
Kadınlarda kusurlu gen, eşleştirilmiş X kromozomu üzerindeki sağlıklı bir gen tarafından telafi edilir, ancak erkeklerde bunu telafi edecek hiçbir şey yoktur. Bu nedenle erkekler, bu hastalıkları annelerinden almalarına rağmen en sık hemofili veya renk körlüğünden muzdariptir.
Bu kadın cinsiyet kromozomunun tamamen beklenmedik bir işleve sahip bir bölgeye sahip olduğu ortaya çıktı: Sperm üretimi için uzmanlaşmış genleri taşıyor.
Bilim insanları, X kromozomunun bilinmeyen fonksiyonunun keşfedilmesinin yanı sıra onun başka bir özelliğini de keşfettiler. Şimdiye kadar Y kromozomunun aksine X kromozomunun stabil olduğuna inanılıyordu. Artık biyologlar onun oldukça hızlı evrimsel değişikliklere uğradığını buldular. Birlikte ele alındığında, bu sonuçlar bizi bunun biyolojik ve tıbbi önemini yeniden düşünmeye zorluyor.
Yukarıda sıralanan hastalıklarla ilişkili olması nedeniyle X kromozomunun çok iyi çalışıldığı düşünülüyordu. Ancak bilim insanları X kromozomunun nükleotid dizisini dikkatli bir şekilde analiz etmeye başladıklarında, daha önce dikkatlerden kaçan detayları keşfettiler. Profesörün laboratuvarı David Sayfası Keşfin gerçekleştiği yer, Y kromozomunun incelenmesi alanındaki çalışmalarıyla biliniyordu. Ve böylece başka bir cinsiyet kromozomunu aldılar ve bu kromozomun insanlarda ve farelerde nükleotid dizisini karşılaştırma görevini üstlendiler. Çalışmanın amacı, uzmanlar arasında X kromozomunun muhafazakar olduğu ve dolayısıyla tüm memelilerde neredeyse aynı olduğu yönündeki köklü görüşü test etmekti.
Doğru bir karşılaştırma yapmak için ekip, Page'in St. Louis'deki Washington Üniversitesi'ndeki araştırmacılarla işbirliği içinde geliştirilen orijinal yöntemini kullanarak insan X kromozomunu yeniden sıraladı. Dizilemenin doğruluğunu geliştirdiler, bu da nükleotid dizisindeki önceden var olan boşlukları doldurmalarına olanak sağladı. Ayrıca araştırmacılar, nükleotid dizisinin aynadaki gibi baş aşağı tekrarlandığı palindrom bölgelerini keşfettiler. Standart yaklaşım bu alanları göz ardı etti. Biyologlar, X kromozomunun rafine yapısını bilim camiasının kullanımına açık hale getirdiler.
Bilim insanları bir karşılaştırma yaptıktan sonra insan ve farelerdeki X kromozomlarının genlerinin yaklaşık %95'ini paylaştığını ve bu genlerin neredeyse tamamının her iki cinsiyette de ifade edildiğini buldu. Bununla birlikte insanları farelerden ayıran 340 gen de keşfedildi. Görünüşe göre farelerin ve insanların ortak atasından bu yana geçen 80 milyon yıl boyunca oluşmuşlar.
Ekspresyonlarının analizi, bu genlerin neredeyse yalnızca sperm üretiminde rol oynadıkları testis hücrelerinde aktif olduğunu ortaya çıkardı.
Çalışmalarını daha iyi anlamak için daha fazla araştırmaya ihtiyaç vardır.
Makalenin ilk yazarı Jakob Müller, "Bu gen grubu tıbbi genetik açısından son derece önemlidir" diyor. - X kromozomunda yer aldıkları için kalıtsal olarak geçmezler. Mendel'in yasaları. Artık konumlarını bulduğumuza göre biyolojik önemlerini analiz etmeye başlayabiliriz."
Bilim insanları bu genlerin erkek üremesi, kısırlık ve muhtemelen testis kanseriyle bağlantılı hastalıklarda önemli bir rol oynadığına inanıyor.
David Page, "X kromozomu insan genomunda en çok çalışılan kromozom olarak kabul edildi" dedi. "Fakat bunun şimdiye kadar bilinmeyen tarafı, hızlı bir şekilde evrimleşmesi ve erkek üreme fonksiyonunda rol alıyor gibi görünmesidir." Sonuçlarımız X kromozomunun ikili rolünü gösterdi. Bu henüz yazılmamış yeni bir kitap."